0. Talktorials
모든 study 는 TeachOpenCADD 를 바탕으로 구성하였습니다.
T006 · Maximum common substructure — TeachOpenCADD 0 documentation
Github:
1. Theory
1-1. Introduction to identification of maximum common substructure in a set of molecules
Maximum Common Structure (MCS)
MCS 는 여러 후보 화합물에서 공통적으로 존재하는 가장 큰 부분 구조를 의미합니다.
maximum common subgraph isomorphism problem 인데, 두 개 이상의 graph 에서 모양이 같고 연결 관계도 같은 subgraph 중 가장 큰 것 (maximum) 을 찾는 문제입니다. (분자 구조는 그래프로 나타낼 수 있는데 이때, 원자는 노드 결합은 엣지로 표현됩니다.)
* node는 점, edge는 선으로 생각하면 쉽게 이해할 수 있습니다.
Cheminformatics 에서 MCS 는 주로 어떻게 활용될까요?
similarity search, hierarchicl clustering, moelcule alignment 등 주로 공통점을 찾는 작업에 많이 활용됩니다.
MCS 의 장점은 직관적인 해석이 가능하다는 것인데, 예를 들어 여러 화합물에서 공통으로 나타나는 구조는 약물 활성에 중요한 부분일 가능성이 높습니다. 구조-활성 관계(SAR) 분석에도 유리합니다. MCS 부분을 강조해서 쉽게 시각화도 가능합니다.
MCS algorithms
알고리즘의 자세한 리뷰는 아래 논문을 참고하시면 좋습니다.
Paper title:
Raymond, John W., and Peter Willett. "Maximum common subgraph isomorphism algorithms for the matching of chemical structures." Journal of computer-aided molecular design 16 (2002): 521-533.

MCS 는 NP-complete problem 입니다. 즉, 두 개 이상의 그래프에서 공통 부분 구조를 찾는 것은 계산적으로 매우 복잡합니다. 이런 문제를 처리하기 위해서는 다양한 유형의 알고리즘을 사용할 수 있는데 보통 Exact algorithms, Approximate algorithms 방식으로 나뉩니다.
Q. 여기서 NP-complete problem 는 무엇일까요..?

알고리즘 이론에서 등장하는 용어라고 합니다. 그림의 용어들부터 정리해보겠습니다. P(Polynomial time)는 문제를 해결하는 빠른 알고리즘이 존재하는 문제들입니다. NP(Non-deterministic Polynomial time)는 정답이 주어진 상태에서는 빠르게 확인 가능하지만, 정답을 직접 찾는 건 보장 안 되는 문제입니다. NP-complete 는 NP 문제 중에서 가장 어렵다고 여겨지는 문제들인데, 모든 NP 문제를 NP-complete 문제로 바꿔서 표현할 수 있습니다. 중요한 것은 한 개라도 P로 풀리면, 모든 NP 문제는 P로 풀립니다. NP-Complete 문제가 P 문제라고 증명이 된다면 P=NP, 하지만 아직 아무도 해결하지 못한 문제입니다.
제가 이해하기로는 이론적으로! NP-complete 중 하나라도 효율적으로 풀리면 나머지 문제도 다 효율적으로 풀 수 있는데 아직까지는 아무도 해결 못한 문제이고, 실전에서는 근사하는 방식으로 결과를 얻는다!
예를 들어, 30개 원자가 있는 분자 2개가 있다. 둘 사이에 겹치는 구조가 있나?
이걸 하나씩 비교해서 맞는 조합을 찾는 건 엄청난 경우의 수가 계산됩니다. (다른거 다 떠나서 2^30 이라고만 계산을 해도 약 10억개의 조합)
Exact algorithms & Approximate algorithms
- Exact: Maximum-clique, backtracking, dynamic programming
- Approximate: Genetic algorithm, combinatorial optimization, fragment storage, …
- (추가) Problem reduction: Simplify the molecular graphs (분자 graph 간소화로 연산량 줄이기)
논문에서는 Exact algorithms & Approximate algorithms 등 알고리즘에 대해서도 구체적으로 설명을 하고 있지만, MCS 를 활용할 때 기본적으로 알아야 할 개념에 대해서만 살펴보겠습니다.

a는 Maximum Common Induced Subgraph (MCIS) 입니다. MCIS 에서 두 그래프는 subgraph of maximum cardinality of vertices (공통적으로 포함되는 가장 많은 수의 점) 을 가지는데, 해당 점에 인접한 모든 edge 도 양쪽 그래프에서 존재해야만 합니다. a 그림에서 G1 과 G2 에서는 점과 연결된 선도 각각 일치한다는 것을 볼 수 있습니다.
b는 Maximum Common Edge Subgraph (MCES) 입니다. MCES 는 이름에서도 알 수 있듯이 두 그래프 간 edge 의 최대 집합을 가진 subgraph 라는 뜻입니다. 점보다는 edge 의 개수에 초점을 둡니다.
G1의 4와 G2의 3' 을 기준으로 MCIS 와 MCES 를 비교해보겠습니다.
a 그림을 나타낸 MCIS 에서는 4와 3'은 매칭되고 있지 않습니다. 그 이유는 G1에서 (2,4) edge 가 존재하는데 G2에서는 3'이 그에 대응하는 edge를 가지고 있지 않기 때문입니다.
b 그림을 나타낸 MCES 에서는 4와 3'이 매칭되고 있습니다.
G1: (3,4), (4,5), (4,7)
G2: (2′,3′), (3′,4′), (3′,7′)
여기서는 eddge 를 기준으로 구조가 맞아 떨어지기 때문에 매칭될 수 있습니다. 점과 점 사이의 연결 여부 보다는 edge 의 패턴을 기준으로 매칭하는 것이 MCES 입니다.
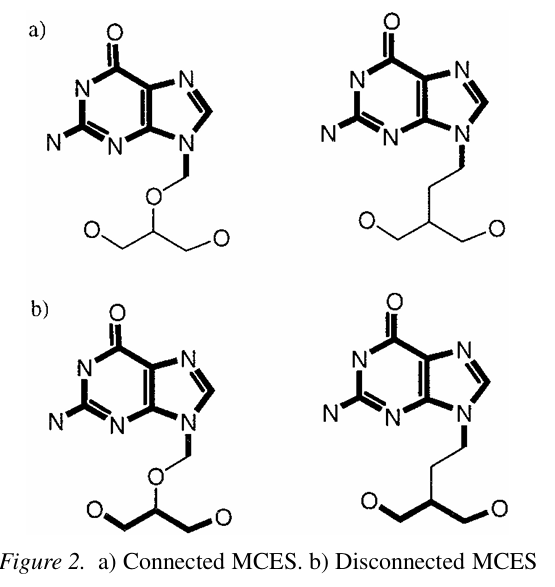
Figure 1. 을 봤을 때는 MCS 가 연결되어 있어야 한다고 오해를 할 수 있지만, 멀리 떨어져 있는 경우에도 MCS 라고 나타낼 수 있습니다. Figure 2. 에서는 a와 b의 예시를 통해 무조건 MCS 가 연결되어야 할 필요는 없음을 보여줍니다. 즉, Connected vs Disconnected MCS는 알고리즘 설계에 큰 영향을 주고(설계 방식, 연산 복잡도가 달라질 수 있습니다.) 이런 연결성 여부는 결과 해석에 영향을 미칩니다. (예를 들어, 같은 크기를 가진 MCS 라도 분리되어 있는 경우에는 화학적으로 덜 유사하다고 판단할 수 있습니다.)
[논문 참고]
- In general, a MCS between a pair of graphs is not necessarily unique as there may be more than one MCS.
- These two classifications can be further divided into those algorithms which are restricted to the case of the connected MCS or are capable of calculating a potentially disconnected MCS.
- It is intuitive that an MCS with fewer disconnected components should greater reflect chemical similarity than one of equal size with more disconnected components.
1-2. Detailed explanation of FMCS algorithm
- Dalke, Andrew, and Janna Hastings. "FMCS: a novel algorithm for the multiple MCS problem." Journal of cheminformatics 5.Suppl 1 (2013): O6.
- RDKit FMCS documentation.
best_substructure = None
pick one structure in the set as query, all other as targets
for each substructure in the query:
convert into a SMARTS string based on the desired match properties
if SMARTS pattern exists in all of the targets:
then it is a common substructure
keep track of the maximum of such substructure
이 방식으로도 MCS 를 찾을 수 있지만, 매우 오래 걸릴 수 있어서 FMCS 에서는 속도를 빠르게 하는 tricks 이 사용됩니다.
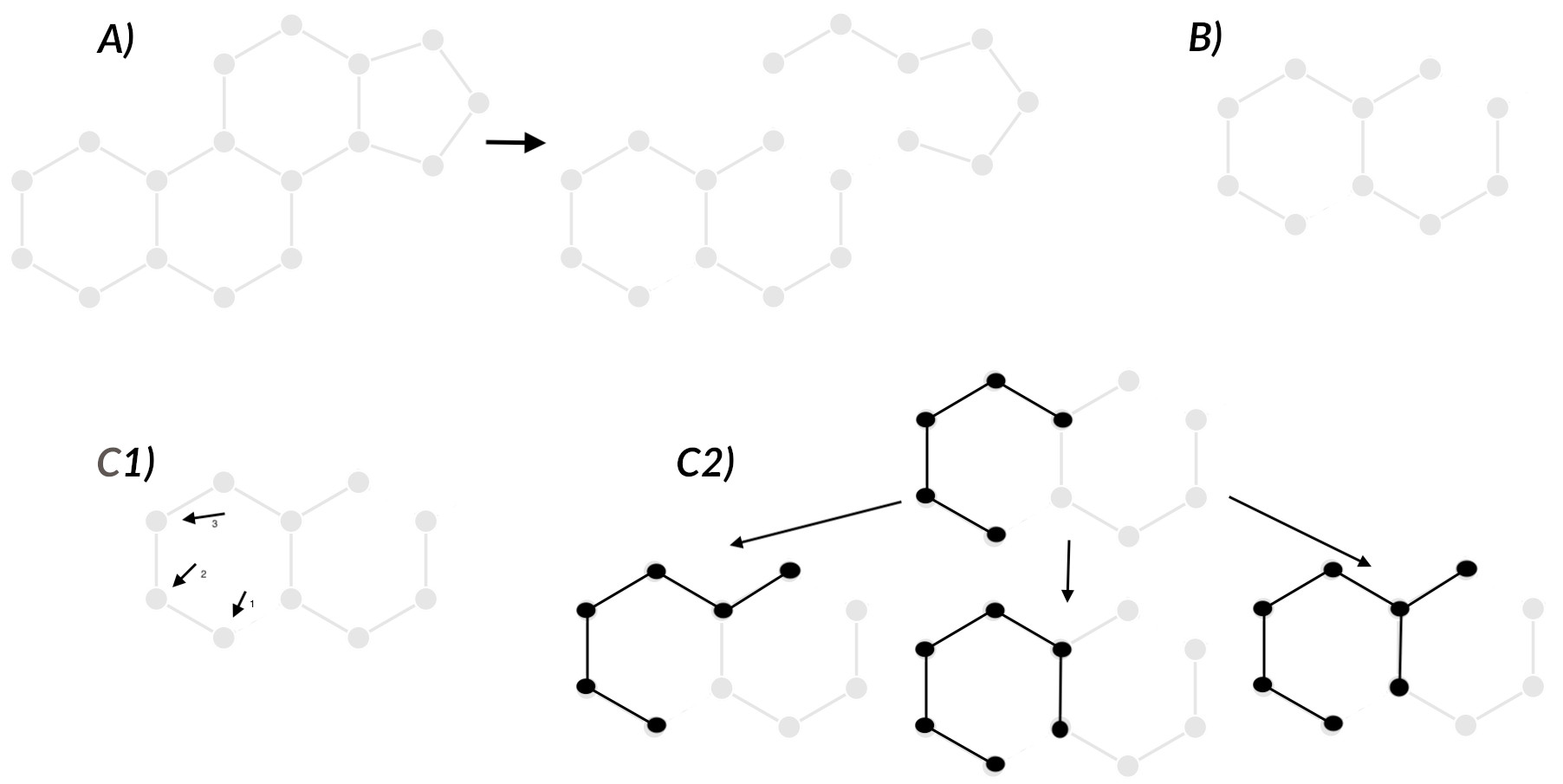
A) 불필요한 Bond 제거
- MCS 에 포함될 수 없는 bond 는 미리 제거
- 모든 input 구조에 atom type 과 bond type 정보가 포함
- Bond type: SMART 로 표현된 (atom1)-(bond)-(atom2)의 형식
- 모든 구조에 공통되지 않는 bond type 은 제외하고 해당 bond(edge) 삭제
- result: atom 정보는 유지되지만 bond 수가 줄어든 graph 생성
B) 가장 단순한 구조를 Query 로 선택
- Heuristic 방식으로 Query 선택
- 각 구조의 가장 큰 fragment 을 찾고, 해당 fragment 의 bond 수를 기준으로 오름차순 정렬
- bond 수가 같으면 원자 수나 입력 순서로 비교
- 가장 작은 fragment 를 가진 구조를 query 로 선택
C를 설명하기 전에, C를 하는 이유는? MCS 의 목적은 공통으로 가지고 있는 subgraph 를 찾는 것입니다. 쉽게 생각하면 subgraph 를 찾기 위해서 subgraph 를 하나씩 만들어보고 그게 모든 분자에 존재하는지 확인하는 방식으로 문제를 해결할 수 있는데, 이런 방식으로 문제를 해결하게 되면 굉장히 오랜 시간이 걸립니다. 그래서 FMCS 에서는 seed (더 작은 단위) 를 기반으로 점점 확장(grow) 시키는 방식으로 진행하여 시간을 단축하고자 합니다!
C1) Seed 기반 subgraph growing
Seed: 현재 fragment graph 에 포함된 atom/bond 정보와 grow 하면 안 되는 bond 정보들 포함 (같은 구조를 여러 번 반복해서 찾지 않기 위해서)
중복 방지를 위해 다음과 같이 seed를 설정:
- 첫 번째 seed: 첫 번째 결합 → 전체 fragment까지 성장 가능
- 두 번째 seed: 두 번째 결합 → 첫 번째 결합 제외
- 세 번째 seed: 세 번째 결합 → 첫/두 번째 결합 제외
C2) Seed growth
- 제외해야 하는 bond 는 사용하지 않고 seed 를 확장시킵니다.
D) 다른 모든 구조에 존재하지 않는 seed 는 제거
- seed growth 후 해당 구조가 모든 분자 구조에 존재하는지 확인하고, 존재하지 않으면 제거
E) growth 가능성 낮은 seed 제거
2. Practical
Getting Started with the RDKit in Python — The RDKit 2025.03.1 documentation
MCS 를 찾을 때 RDKit 을 많이 이용하기 때문에 RDKit 에서 제공하는 documentation 기반해서 설명드리겠습니다.
RDKit 에서는 MCS 를 찾는 두 종류의 방법이 있습니다.
(1) FindMCS
- Finds a single fragment maximum common substructure (MCS) of two or more molecules
- 앞의 논문에서 MCS 는 분자내에서 떨어진 구조여도 MCS 가 될 수 있다고 하였습니다. 하지만, FMCS 는 단 하나의 MCS 를 찾는 것이 특징입니다.
(2) RascalMCES
- Finds the maximum common edge substructure (MCES) between two molecules and can return a multi-fragment MCES.
- RascalMCES 같은 경우에는 2개 이상의 MCES 를 찾을 수 있는 것이 특징입니다.
두 방식의 차이는 그림을 통해서 보면 바로 알 수 있습니다.
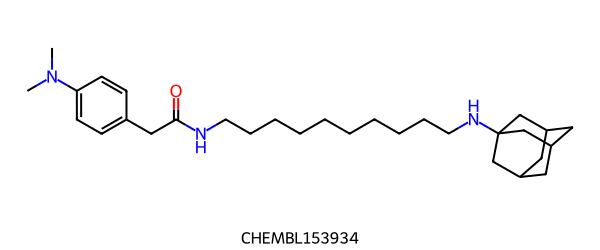

두 분자가 있습니다.
FMCS:

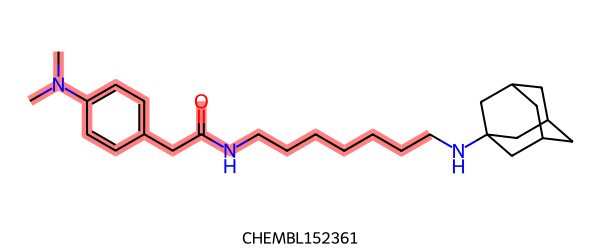
FMCS 는 single fragment 를 찾습니다.
RascalMCES:


RascalMCES 는 2개 이상의 MCES 를 찾습니다.
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit.Chem import rdFMCS
from IPython.display import display
# 분자 리스트
mols = [
Chem.MolFromSmiles("O=C(NCc1cc(OC)c(O)cc1)CCCC/C=C/C(C)C"),
Chem.MolFromSmiles("CC(C)CCCCCC(=O)NCC1=CC(=C(C=C1)O)OC")
]
# 조건 다르게!
mcs1 = rdFMCS.FindMCS(mols, bondCompare=rdFMCS.BondCompare.CompareAny)
mcs2 = rdFMCS.FindMCS(mols, bondCompare=rdFMCS.BondCompare.CompareOrderExact)
mcs_mol1 = Chem.MolFromSmarts(mcs1.smartsString)
mcs_mol2 = Chem.MolFromSmarts(mcs2.smartsString)
def get_highlights(mol_list, mcs_mol):
return [mol.GetSubstructMatch(mcs_mol) for mol in mol_list]
highlight_atoms_1 = get_highlights(mols, mcs_mol1)
highlight_atoms_2 = get_highlights(mols, mcs_mol2)
img1 = Draw.MolsToGridImage(
mols,
highlightAtomLists=highlight_atoms_1,
molsPerRow=2,
subImgSize=(300, 300),
legends=["Mol1", "Mol2"],
useSVG=False
)
img2 = Draw.MolsToGridImage(
mols,
highlightAtomLists=highlight_atoms_2,
molsPerRow=2,
subImgSize=(300, 300),
legends=["Mol1", "Mol2"],
useSVG=False
)
display(img1) # bondCompare=CompareAny
display(img2) # bondCompare=CompareOrderExact
FMCS 에서는 MCS 를 찾는 Options 을 지정할 수 있습니다. 코드처럼 bond 를 기준으로 옵션을 바꿀 수 있습니다. (원자를 기준으로도 가능합니다.)

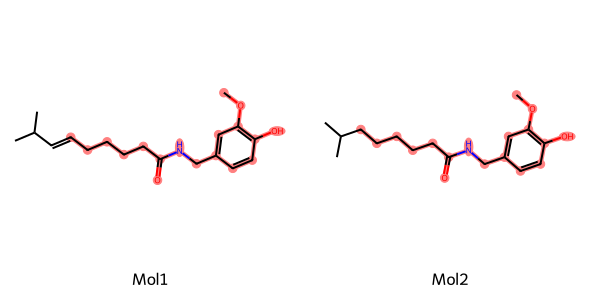
첫 번째 조건보다 두 번째 조건에서 좀 더 기준이 strict 한 것을 알 수 있습니다.
Reference
- Raymond, John W., and Peter Willett. "Maximum common subgraph isomorphism algorithms for the matching of chemical structures." Journal of computer-aided molecular design 16 (2002): 521-533.
- NP-완전 - 위키백과, 우리 모두의 백과사전
- Dalke, Andrew, and Janna Hastings. "FMCS: a novel algorithm for the multiple MCS problem." Journal of cheminformatics 5.Suppl 1 (2013): O6.
- RDKit FMCS documentation.
- Getting Started with the RDKit in Python — The RDKit 2025.03.1 documentation
- 현재 관련 분야를 공부하고 있는 전문가가 아닌 학생이기 때문에 틀린 내용이 있을 수 있습니다.
- 오타와 틀린 부분을 댓글로 알려주시면 수정하도록 하겠습니다.
'Drug study > TeachOpenCADD' 카테고리의 다른 글
| T008 · Protein data acquisition: Protein Data Bank (PDB) (4) | 2025.07.03 |
|---|---|
| T007 · Ligand-based screening: machine learning (2) | 2025.05.13 |
| T005 · Compound clustering (0) | 2025.04.18 |
| T004 · Ligand-based screening: compound similarity (2) | 2025.04.16 |
| T003 · Molecular filtering: unwanted substructures (0) | 2025.04.08 |